Vad är Retinis Pigmentosa (RP)?
RP, Retinitis pigmentosa, är ett samlingsnamn på ett hundratal ärftliga ögonsjukdomar som drabbar ögats näthinna. Sjukdomen och graden av synskada varierar avsevärt mellan olika familjer men också inom samma familj. Man tror att det finns omkring 4000 personer med ärftlig näthinnesjukdom i Sverige. RP är den vanligaste orsaken till grav synskada i yrkesverksam ålder.
Symton Retinitis är latin och betyder inflammation av retina, det vill säga näthinnan. Termen är gammal och felaktig eftersom RP inte är någon inflammation utan en degeneration. Med degenerativ menas att det sker en fortlöpande förstörelse av celler. I detta fall förstörs ögats synceller, det vill säga tappar och stavar. Ordet pigmentosa kommer av att det hos de flesta med RP bildas pigmentklumpar i ögonbotten. Symtomen kan variera men de synproblem som upptäcks först är i regel:
- Nedsatt förmåga att se i svagt ljus och mörker. Detta kallas för nattblindhet. Det är stavarna som vi ser med i skymningen och det är oftast deras funktion som först försämras.
- Ökad bländingsbenägenhet. Ögat har allt svårare att ställa om sig från ljus till mörker och tvärtom.
- Senare kan också synfältet inskränkas så att man får svårt att se åt sidorna.
- Tapparna, de synceller som styr vårt färg-, dag- och detaljseende, förstörs i regel efter stavarna. Då får man först små fläckar med dålig syn i synfältet. Dessa fläckar blir med tiden större.
- Till slut kanske bara ett kikarseende återstår. Då har sidoseendet försvunnit och det är bara en liten del av det centrala seendet som finns kvar.
- Synförlusten kan till sist resultera i att enbart så kallat ljus- och mörkerseende återstår. Ledsynen har gått förlorad.
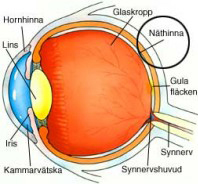
Köp boken "Att leva med RP" och stöd ögonforskning på retinitis pigmentosa

Att leva med RP är en handbok för personer med RP, anhöriga och den övriga omgivningen. Boken vill ge en bred beskrivning av Retinitis Pigmentosa, från forskning till hur det kan vara att leva, arbeta och utvecklas med en degenerativ ögonsjukdom.
I boken finns också praktiska råd och tips om hur det lättare kan gå att hantera de situationer som uppstår, för såväl personer med RP som för anhöriga, kollegor och samhället i övrigt.
Boken kostar 150 kronor för medlemmar i Svenska RP-föreningen och 200 kronor för övriga. Beställer du fler än 10 böcker gäller medlemspris.
75 kronor av bokpriset går till RP-forskning.
Du köper RP-boken genom att sätta in bokpriset plus porto 40 kronor (Vid beställning av 10 eller fler böcker kostar portot 100 kronor), på RP-föreningens plusgiro 62 21 08-9. Märk talongen RP-boken.
Om du vill bli medlem i Svenska RP-föreningen eller har frågor kring boken kontaktar du RP-föreningens kansli på telefon 08-702 19 02 säkrast klockan 9-13 eller via e-post adm@srpf.a.se
Svenska RP-föreningen
Prognos och behandling
RP kan vara både svår och lätt att upptäcka. Sjukdomen kan skada synen redan i barndomen medan andra har en lätt synnedsättning först på åldern höst. Ärftlighet, symtom och prognos varierar avsevärt mellan olika RP-typer. Klassisk RP är lätt att diagnostisera på grund av att ögonbotten får karaktäristiska pigmentklumpar. Hos små barn finns det dock inga pigmentklumpar så då tas ett ERG, elektroretinogram. ERG är en undersökningsmetod som liknar EKG. En tunn elektrod placeras i en kontaktlins som mäter den ström som bildas när ögat utsätts för ljus. Hos dem med RP förekommer mycket svaga elektriska signaler. Ofta är den kurva som mäts helt utslätad. Detta går att se redan innan barnet fått ett försämrat mörkerseende. En annan viktig undersökningsmetod är fotografering av ögonbotten. Man följer sjukdomsförloppet med funktionstester som ERG och synfältsundersökningar. En ögonläkare kan oftast konstatera RP. Men för att veta exakt vilken RP-typ det rör sig om behövs en specialistutredning. En sådan undersökning kan oftast ske vid ögonklinikerna vid våra universitetssjukhus. För att få en sådan undersökning kan det ofta vara nödvändigt att söka vård utanför det egna landstinget. Att få rätt diagnos är viktigt eftersom sjukdomens utveckling kan variera högst väsentligt. Vissa RP-typer ger grav synskada medan andra är mer stabila. Någon effektiv behandling av RP-sjukdomarna finns ännu inte. Resultaten från en tidigare amerikansk studie har visat att synnedgången hos vissa RP-former möjligen kan bromsas med A-vitaminer. Därför ges doser av A-vitamin i kapslar eller droppar till somliga med RP. Doserna är inte så stora, 15 000 IE per dag, men man är försiktigt vid behandling av gravida och leversjuka. Barn behandlas enbart efter samråd med en barnläkare. En anledning till att försöka få en korrekt diagnos är sjukdomens ärftlighet. Att fastställa hur sjukdomen går i arv kan bland annat ha betydelse när det gäller att fastställa hur omfattande synskadan kommer att bli. Med hjälp av kombination av ERG, synundersökning och DNA-test kan man idag lättare klarlägga vilken form av RP som föreligger. Vid en genundersökning lämnar en person med RP eller en frisk anlagsbärare ett blodprov. I långt ifrån alla fall går det inte att få besked om vilken RP-typ man har och ej heller besked om hur omfattande synskadan kommer att bli. I Lund har man med hjälp av svenska och utländska kollegor klarlagt ett stort antal gendefekter hos cirka 300 svenska familjer med olika former av RP.
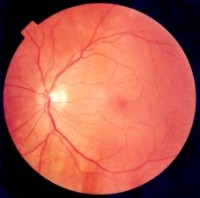
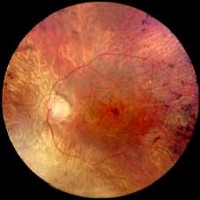
Orsak och förebyggande åtgärder
Man brukar säga att anledningen till RP är störd ämnesomsättning i ögats näthinna. Det kan vara:
- Fel på de proteiner som deltar i processen att omvandla ljuset till en nervsignal.
- Fel på de proteiner som bygger upp strukturen på tappar och stavar.
- Eller så kan det vara fel på de enzymer som har att göra med cellens ämnesomsättning.
Det finns alltså tre olika processer i ögats celler som det kan vara fel på.
Rehabilitering Rehabilitering är mycket viktig om sjukdomen gett upphov till nedsatt syn och lässvårigheter. Målet är att alla ska kunna leva ett oberoende liv. Syncentralen i Sverige har ögonläkare, optiker, synpedagog och erbjuder olika hjälpmedel. Samhället ger stöd åt personer med funktionshinder. Graden av funktionshinder avgör vilken typ av stöd och service man är berättigad till. Kommunen ansvarar för stöd som kan underlätta vardagen för personer med funktionshinder, som till exempel assistans och hjälp i hemmet. Landstinget har ansvar för hälso- och sjukvård, samt rehabilitering och hjälpmedel. Försäkringskassan handlägger och beviljar ekonomiskt stöd för den i arbetsför ålder.
Forskning RP-forskningen är mycket intensiv och världsomspännande. Huvudspåren inom forskningen kan mycket kort beskrivas såhär:
- Transplantation av synceller och stamceller.
- Att med genteknik fastställa var felet i ämnesomsättningen sitter.
- Behandling med genterapi som skulle kunna vara en möjlig bot.
- Att med mediciner försöka stoppa eller bromsa celldöden, förstörelsen av de ljuskänsliga syncellerna och tapparna.
- Rp-registret innefattande 2800 patienter är knutet till speicalmottagningen i Lund.
I Lund pågår sedan många år djurförsök med transplantationer av näthinneceller. Mekanismen bakom den celldöd som sker i ögat hos de flesta personer med RP sker möjligen genom apoptos - förutbestämd celldöd. En möjlig behandling i framtiden skulle då också kunna vara medicinering som stoppar celldöden. Det finns flera sorters djur som har ögonsjukdomar som motsvarar RP hos människor, till exempel katt och hund. På dessa djur utförs mycket forskning för att man ska förstå mer av sjukdomen. Genom internationellt samarbete försöker forskare hitta den genetiska defekten som finns hos varje enskild familj med RP. Denna forskning, som bland annat sker i Umeå och Lund är mycket viktigt då det på sikt kan leda till en behandling. Aktuell forskning med genterapi har nu visat sig så lovande att patienter med RP ingår i dessa forskningsstudier i och nyligen presenterades 2 års uppföljning av 12 behandlade patienter i USA. Vid avdelningen för oftalmologi i Lund finns samarbete med USA för planerade genterapi vid en speciell form, som kallas kongenital retinoschis.
Texten om RP är faktagranskad av Professor Sten Andréasson, Överläkare på Lunds Universitetssjukhus